MISSION
To accelerate the transition to a renewable energy society by discovering new materials, chemicals, and processes through multi-scale simulation and data science.

RESEARCH
We interface multi-scale materials simulation and data science. Specifically, we develop innovative methods that accelerate materials design.
Latest Publications
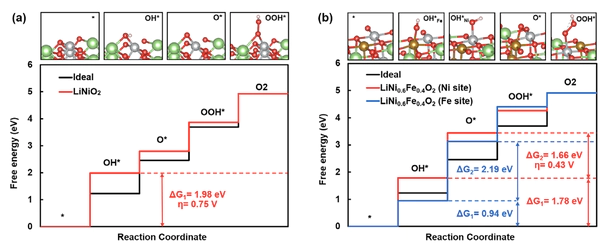
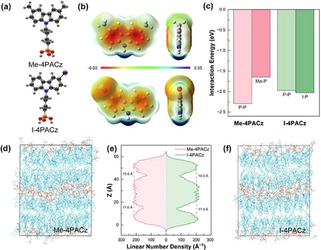
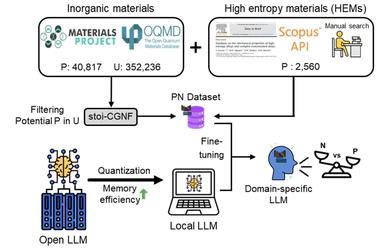
Latest News and Gallery
Happy Teacher’s Day
To mark Teacher’s Day, META Lab prepared a small event for our professor.
Beyond the research, he is someone we genuinely look up to as a person. We are grateful to learn from him every day.
Happy Teacher’s Day! 🌹
P.S. The cake was enjoyed by the grad students themselves. 🎂
P.S. The cake was enjoyed by the grad students themselves. 🎂

4 Prizes in One Conference
META Lab swept the 2026 KSIEC Spring Conference, bringing home four awards — a testament to the talent of our students and the mentorship of our professor. This is a proud demonstration of META Lab’s research excellence.
Congratulations to Daeyeong (Oral) and Jiwoo, Youngtak, and Taegyeong (Poster) on their well-deserved awards! 🏆

META Lab at the 2026 KSIEC Spring Conference
META Lab members attended the 2026 Spring Meeting of the Korean Society of Industrial and Engineering Chemistry (KSIEC), presenting our latest research and engaging in meaningful academic exchanges with fellow researchers from across the field.
Through these interactions, our members gained fresh perspectives and new insights, contributing to META Lab’s continued growth as a research group.
