MultiScale Simulation Strategy
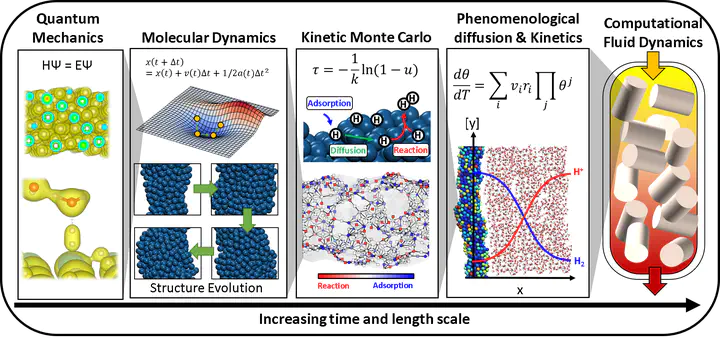
The time and length scale at which chemical phenomena occur can be categorized into several types. At the smallest scale that catalysis scientists consider, electrons rapidly equilibrate their orbital given the position of their nucleus which occurs at a time less than a picosecond. These atoms can vibrate around their bond length and molecules move around the space which constitutes thermodynamic properties that we calculate using statistical thermodynamics. These vibrating, translating, and rotating molecules can react together to form new molecules occurring at a time scale of about a picosecond. This set of reactions then changes the local concentration within, for example, a reactor at a time scale of a second. At a larger scale, the concentration gradient within a reactor needs to be accounted for via convection and diffusion. To account for these various phenomena that obey different physical laws, we need to use several tools in order to simulate the observable properties such as conversion, selectivity, etc.
Quantum mechanics simulation
Molecular dynamics
At a larger scale, atoms in a molecule vibrate around their equilibrium position. The molecules rotate and translate freely in the case of gas and liquid states. DFT is usually used as a tool to calculate the ground state of the system. For example, water molecules form hexagonal layers when it becomes ice. However, as the temperature rises, the ice melts and the water structure becomes irregular. For these high entropy states, which is a system with a very large number of structural configurations with similar energy, calculating the ground state structure is not sufficient, but the motions of atoms need to be explicitly accounted for to understand the emergent properties of the high entropy system. The molecular dynamics simulates and tracks the individual motions of atoms at each time state, thus we can understand the emergent properties. See these great simulation videos done by scientists (video1, video2) for examples of water molecular dynamics simulation.
Kinetic Monte Carlo and phenomenological kinetics
Computational fluid dynamics
The placement of the catalyst pallets in the reactor, the fluid characteristics, and the mixing affect the concentration gradient that exists in the reactor. While all catalyst pallets follow the kinetics based on their quantum mechanical characteristics, statistical thermodynamics, and kinetics, the gradients created by the fluid characteristics in the reactor create the concentration gradients, which change not just based on the kinetics, but also based on the diffusion and convection. Thus, to understand the final products of a reactor system that you would observe in the chemical plant, the fluid dynamics are simulated. See this video created by CPFD software.