Practical Deep-Learning Representation for Fast Heterogeneous Catalyst Screening
Abstract
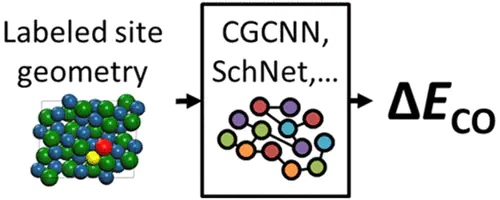
The binding site and energy is an invaluable descriptor in high-throughput screening of catalysts, as it is accessible and correlates with the activity and selectivity. Recently, comprehensive binding energy prediction machine-learning models have been demonstrated and promise to accelerate the catalyst screening. Here, we present a simple and versatile representation, applicable to any deep-learning models, to further accelerate such process. Our approach involves labeling the binding site atoms of the unrelaxed bare surface geometry; hence, for the model application, density functional theory calculations can be completely removed if the optimized bulk structure is available as is the case when using the Materials Project database. In addition, we present ensemble learning, where a set of predictions is used together to form a predictive distribution that reduces the model bias. We apply the labeled site approach and ensemble to crystal graph convolutional neural network and the ∼40 000 data set of alloy catalysts for CO2 reduction. The proposed model applied to the data set of unrelaxed structures shows 0.116 and 0.085 eV mean absolute error, respectively, for CO and H binding energy, better than the best method (0.13 and 0.13 eV) in the literature that requires costly geometry relaxations. The analysis of the model parameters demonstrates that the model can effectively learn the chemical information related to the binding site.