Progress in Computational and Machine-Learning Methods for Heterogeneous Small-Molecule Activation
Abstract
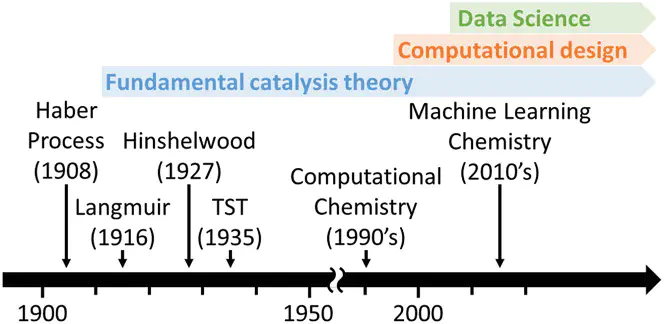
The chemical conversion of small molecules such as H2, H2O, O2, N2, CO2, and CH4 to energy and chemicals is critical for a sustainable energy future. However, the high chemical stability of these molecules poses grand challenges to the practical implementation of these processes. In this regard, computational approaches such as density functional theory, microkinetic modeling, data science, and machine learning have guided the rational design of catalysts by elucidating mechanistic insights, identifying active sites, and predicting catalytic activity. Here, the theory and methodologies for heterogeneous catalysis and their applications for small-molecule activation are reviewed. An overview of fundamental theory and key computational methods for designing catalysts, including the emerging data science techniques in particular, is given. Applications of these methods for finding efficient heterogeneous catalysts for the activation of the aforementioned small molecules are then surveyed. Finally, promising directions of the computational catalysis field for further outlooks are discussed, focusing on the challenges and opportunities for new methods.