Structure-Based Synthesizability Prediction of Crystals Using Partially Supervised Learning
Abstract
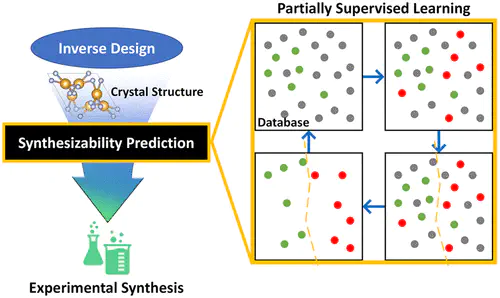
Predicting the synthesizability of inorganic materials is one of the major challenges in accelerated material discovery. A widely employed approximate approach is to consider the thermodynamic decomposition stability due to its simplicity of computing, but it is notorious for either producing too many candidates or missing important metastable materials. These results, however, are not unexcepted since the synthesizability is a complex phenomenon, and the thermodynamic stability is just one contributor. Here, we suggest a machine-learning model to quantify the probability of synthesis based on the partially supervised learning of materials database. We adapted the positive and unlabeled machine learning (PU learning) by implementing the graph convolutional neural network as a classifier in which the model outputs crystal-likeness scores (CLscore). The model shows 87.4% true positive (CLscore > 0.5) prediction accuracy for the test set of experimentally reported cases (9356 materials) in the Materials Project. We further validated the model by predicting the synthesizability of newly reported experimental materials in the last 5 years (2015–2019) with an 86.2% true positive rate using the model trained with the database as of the end of year 2014. Our analysis shows that our model captures the structural motif for synthesizability beyond what is possible by Ehull. We find that 71 materials among the top 100 high-scoring virtual materials have indeed been previously synthesized in the literature. With the proposed data-driven metric of the crystal-likeness score, high-throughput virtual screenings and generative models can benefit significantly by effectively reducing the chemical space that needs to be explored experimentally in the future toward more rational materials design.