Group Additivity for Thermochemical Property Estimation of Lignin Monomers on Pt(111)
Abstract
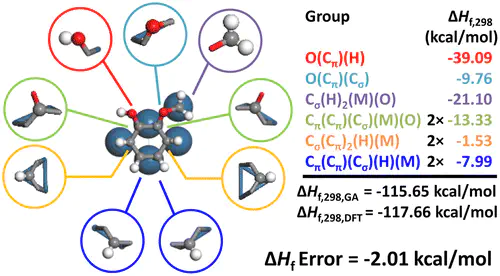
Conversion of biomass has received considerable attention, but theoretical investigation is challenging due to the large computational burden. In this contribution, we explore group additivity for computing the thermochemistry of lignin monomers, and by extension to single-ring aromatic hydrocarbons, on Pt(111). We find that the previous framework developed for open-chain molecules and furanics is inadequate for lignin monomers due to conjugation. Using quantum theory of atoms in molecules (QTAIM), we find that the type of binding of the adsorbate atoms with the surface sites, for example, σ, σσ, or σπ, has an important impact on the conjugation of bonds in adsorbates. We introduce two new models that account for the type of binding of the central atom and its nearest neighbors, namely, a conjugation-based and a site-based scheme, which result in significant improvement. A total of 591 density functional theory data points were regressed; cross-validation of the site-based scheme reveals that mean absolute errors are 2.81 kcal/mol in ΔHf,298, 1.07 cal/(mol K) in ΔS298, and 0.25 cal/(mol K) in Cp,300. The slightly simpler conjugation-based model, which does not resolve the binding type of all nearest neighbors, also performs well.