Strongly Coupled Metal/Amorphous Ru/RuOx Heterostructure for Efficient Electrocatalytic Hydrogen Production
Abstract
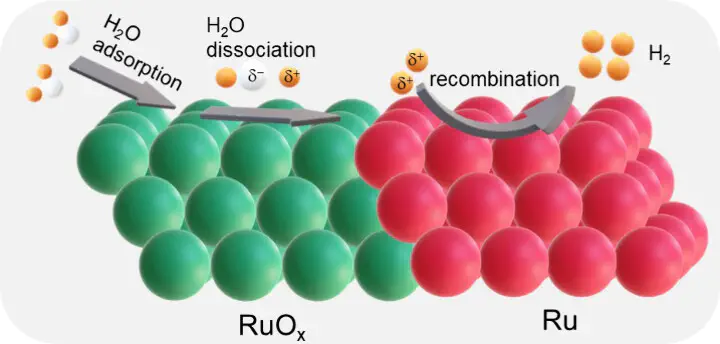
Constructing well-defined heterostructure interfaces in catalysts is an effective strategy to break scaling relationships and accelerate reactions involving multiple intermediates. In this study, a heterostructure catalyst consisting of crystalline ruthenium (Ru) and amorphous ruthenium oxide (RuOx) nanoparticles uniformly distributed on N-doped carbon was developed by using a low-temperature (500 °C) pyrolysis method. The strong and well-defined electronic interactions at the interface between Ru and RuOx synergistically optimize hydrogen adsorption and desorption at the heterointerfaces of each particle, thereby significantly accelerating the kinetics of the hydrogen evolution reaction (HER). Consequently, the synthesized catalysts achieve pH-universal HER performance and exhibit impressively low overpotentials of 11 mV to reach a current density of 10 mA cm–2 under alkaline conditions. Additionally, the anion exchange membrane electrolyzer delivers remarkably low voltages of 2.08 V to achieve a current density of 0.82 A cm–2 and demonstrates prolonged stability over 100 h at a current density of 500 mA cm–2, outperforming Pt/C catalysts. Density functional theory calculations further reveal that amorphous RuOx effectively reduces the free energy barrier of the water dissociation step, while adjacent Ru promotes hydrogen evolution. This synthesis strategy offers a viable approach for the rational design and synthesis of superior HER electrocatalysts.