Integrating reaction enumeration and machine learning for microkinetic modeling of bond-exchange reactions
Abstract
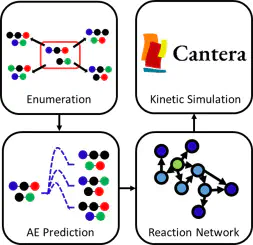
An automated rapid microkinetic simulation is invaluable in predicting the products of chemical reactions ahead of experiments. Although existing models perform well in their focused domains, a dedicated comprehensive framework for the bond exchange reaction kinetics is absent. To address this, we present an integrated algorithm for the rapid kinetic simulation of bond exchange reaction networks. We introduce a scalable matrix-based enumeration method that allows economic exploration of all plausible reaction products without resorting to reaction templates. Our model primarily uses machine learning, which achieves a mean absolute error of 4.55 kcal/mol for activation energies, to more efficiently predict reaction properties, without relying on stored chemical reaction databases or expensive electronic structure calculations. The framework was validated by successfully reproducing two reactions.