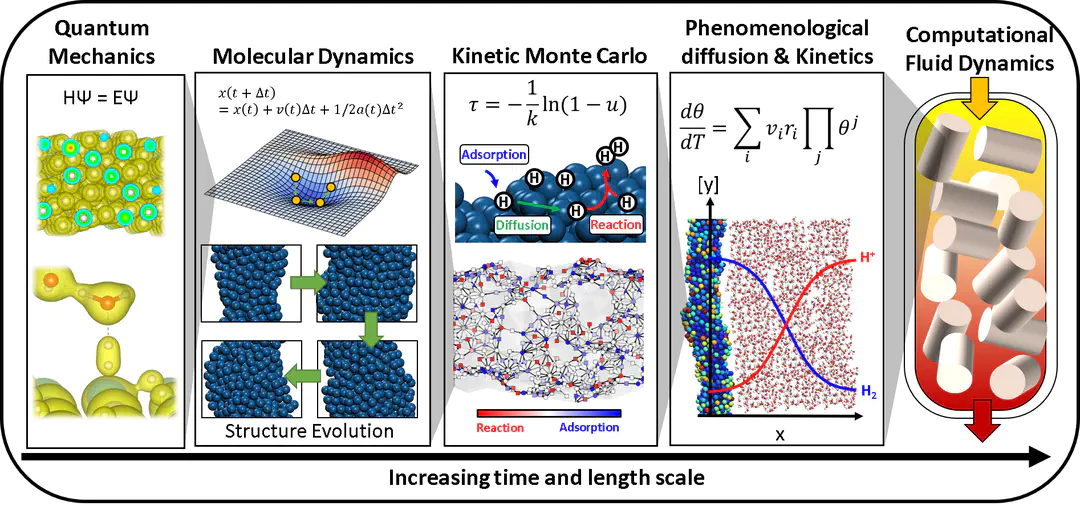
RESEARCH APPROACH
The fundamental properties of materials are determined by electronic interaction between atoms. The advancement of density functional theory (DFT) has enabled the electronic structure calculations, but DFT cannot directly calculate the observable properties (e.g. conversion, and selectivity) as the time and length scale of the observable phenomena is about 12-15 orders of magnitude larger than the scale of the system that can be simulated using DFT. To enable the simulation of observable properties, we use an arsenal of statistical thermodynamics-based tools, where each tool is specialized in a specific time and length scale. By coupling each scales' calculation, we go beyond the electronic properties and calculate observables, enabling us to validate our model and predictively optimize our materials. See ref1, and ref2.
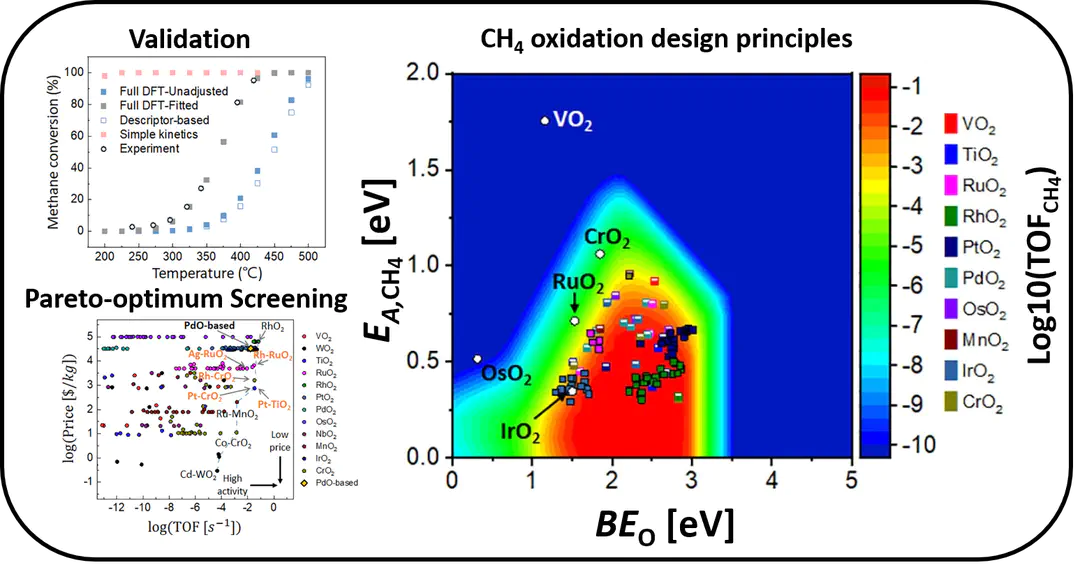
With the multiscale simulation strategy, we can elucidate a powerful design princples for processes. For example, the graph on the left demonstrates the turnover frequency of thermal CH4 emission reduction to CO2. We found that oxygen atom's binding energy and CH4 molecules' activation energy can describe the metal oxides' reduction activity. This plot explains activity for various mono metal oxides as plotted, and, by computing these energies for various materials (so called high-throughput screening), we can discover potential new active materials. Our research involves elucidating these design principles for the process and system of interest and performing high-throughput screening. see ref.
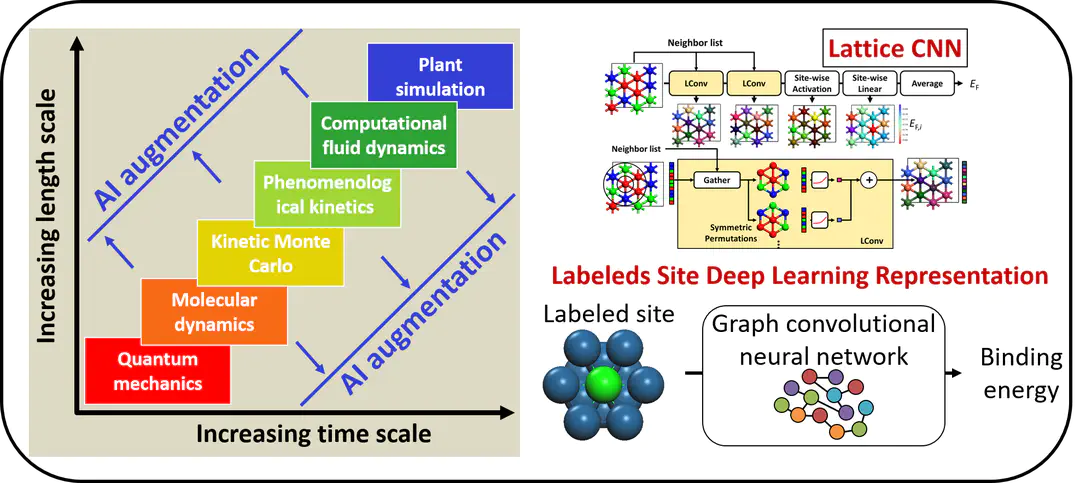
Even with the multiscale simulation strategy, our simulation capacity is limited by the expensive quantum mechanics calculations. With the US materials genome initiative and advancement in artificial intelligence, we have seen rapid growth in data science in the past decade. The flexibility of the machine learning models allows us to simulate systems that were not been possible before. We have one of the leading-edge experiences in developing deep learning models that have enabled us to look at the unprecedently complex system, expanding our multiscale simulation capability. Our current research focuses on developing deep learning methods that will innovate the materials design paradigm. See ref1, ref2, and ref3.
Key Expertise
Materials
Transition metal catalyst
Single atom catalyst
Graphene
Metal oxides
Metal-organic frameworks
Perovskite

Objectives
New materials discovery
Atomistic mechanism elucidation and design
Interface engineering
Uncertainty quantification
Synthesizability prediction
Techniques
Deep learning
Microkinetic modeling
Density functional theory
Force field
Generative models
Unsupervised learning
Large reaction network modeling
Technology
Electrochemistry
Reforming catalysis
Biomass conversion
Shale gas utilization
Photoactive materials
Reaction safety